协“爱”共进,一起向未来
上海交通大学医学院附属新华医院
戴选彤 姚陈琳 林芙君
谈及遗传性肾炎,大家往往谈虎色变,今天就让我们走进一种典型遗传性肾炎――Alport综合征(alport syndrome, AS)的世界。相信除了从事罕见病专业的医务人员以及部分患友,大部分人对于此类疾病的认知相当有限,特别是因为其属于罕见病以至于大众总觉得发生概率极低与我无关,但其实AS在人群中并不罕见。目前AS的人群患病率据估算可占到成人慢性肾脏病(chronic kidney disease,CKD)的3%,而根据柳叶刀2020年发表的大型流行病学统计提示2017年全球CKD平均患病率为9.1%(共约6.975亿),因此粗略估计全球约2000万AS患者。
什么是Alport综合征
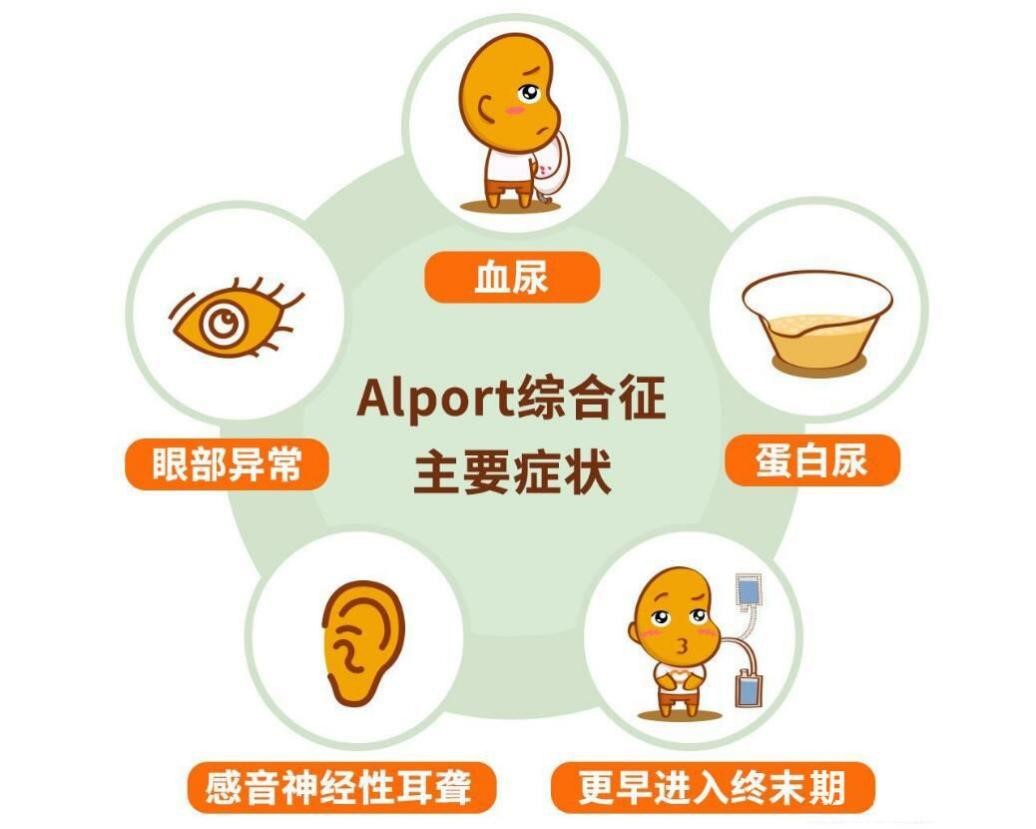
Alport综合征(Alport syndrome,AS)是临床上相对常见的遗传性肾小球疾病,并被列为2018年国家卫生健康委颁布的首批121种罕见病目录。AS以肾性血尿、蛋白尿和进行性肾功能减退为临床特点,可合并感音神经性耳聋及眼部异常等肾外症状,肾活检可见肾小球基底膜(glomerular basement membrane,GBM)发生弥漫变薄、厚薄不均、致密层撕裂或分层或虫蚀样等改变,部分合并局灶节段性肾小球硬化(focal segmental glomerulosclerosis, FSGS)。对于AS患者,肾脏会逐渐失去过滤血液中废物的能力。在疾病后期,患者不得不需要进行慢性透析治疗或肾移植。鉴于目前大众群体对遗传性肾脏病的了解不多,其中一些患病者在诊疗初期往往都经历过恐惧、紧张、焦虑、迷茫、逃避等心理阶段,一些患者及家庭往往因恐惧,内心逃避面对疾病而错失最佳治疗时机。甚至少部分家庭盲目信任所谓偏方花费大量时间和金钱,导致最终“家破人亡”。
确诊为Alport综合征后如何治疗?
首先,确诊为AS后患者及其家属应积极调整心态并及时联系专科医师,不恐惧不逃避,配合专科医师进行疾病评估、家系谱调查及药物治疗等。不同类型的AS预后差异较大,部分患者可保持终身血尿蛋白尿。
对于AS的治疗,目前仍以肾素-血管紧张素系统抑制剂(Renin-angiotensin system inhibitors,RASI)为主,虽不能完全阻止或逆转肾功能进展,但早期用药不仅可以延长生存期、提高生存质量,也能在一定程度上减轻费用负担。而一旦拖延治疗,部分AS患者最终会发展成尿毒症,就只能依靠透析和肾移植来维系生命。虽然相对于其他肾脏疾病,AS患者肾移植后复发概率较低,但肾源和高额医疗费用也使不少家庭望而却步。
新药研发――众人拾柴火焰高
目前关于AS新药相关临床研究正如火如荼的开展,有以抗肾脏纤维化和/或抗氧化及炎症调节为主要靶点的miRNA-21抑制剂Lademirsen和核因子E2相关因子2(nuclear factor E2-related factor 2,Nrf2)激活剂Bardoxolone methyl(bardoxolone)、选择性内皮素受体拮抗剂Atrasentan、钠-葡萄糖协同转运蛋白2抑制剂(sodium-dependent glucose transporters 2,SGLT-2)、酮嗪片、熊去氧胆酸、羟氯喹等。
另外关于AS小鼠模型的研究提供了的许多潜在的细胞信号靶点可在未来成为新药研发方向。而基因编辑―治疗、干细胞移植等也是目前热门研究方向,未来可期。
协“爱”共进,一起向未来
相信在国家的有利政策,医务人员、科学家、医药企业的共同努力下,未来在新药研究领域会有源源不断的捷报传来,使更多的AS患者获得安全有效的诊疗,减轻患者本人及其家庭对于疾病带来的生理、心理的痛苦及沉重的经济负担。