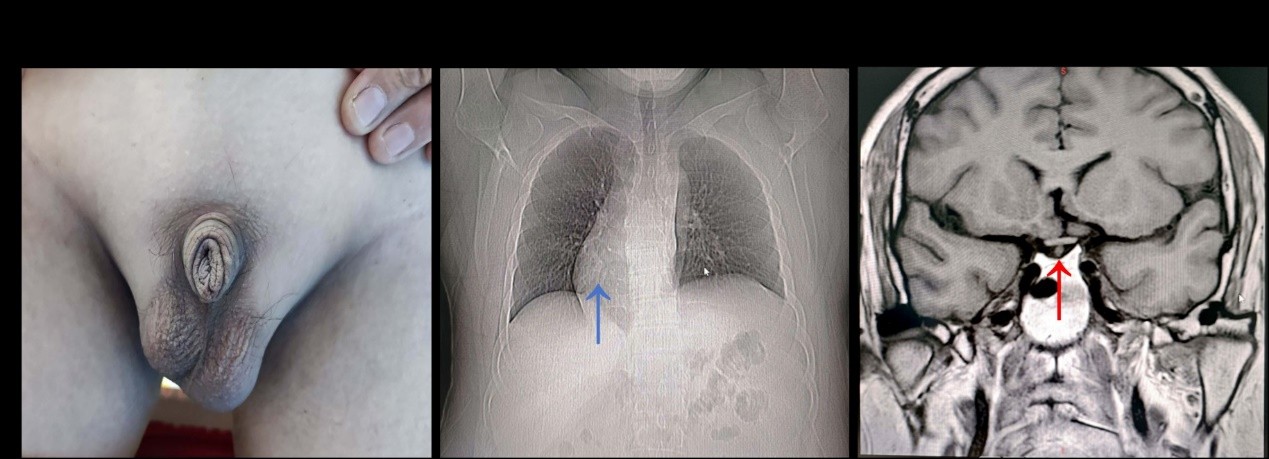
�������ۺ����ϲ�ԭ����Ĥ����1��
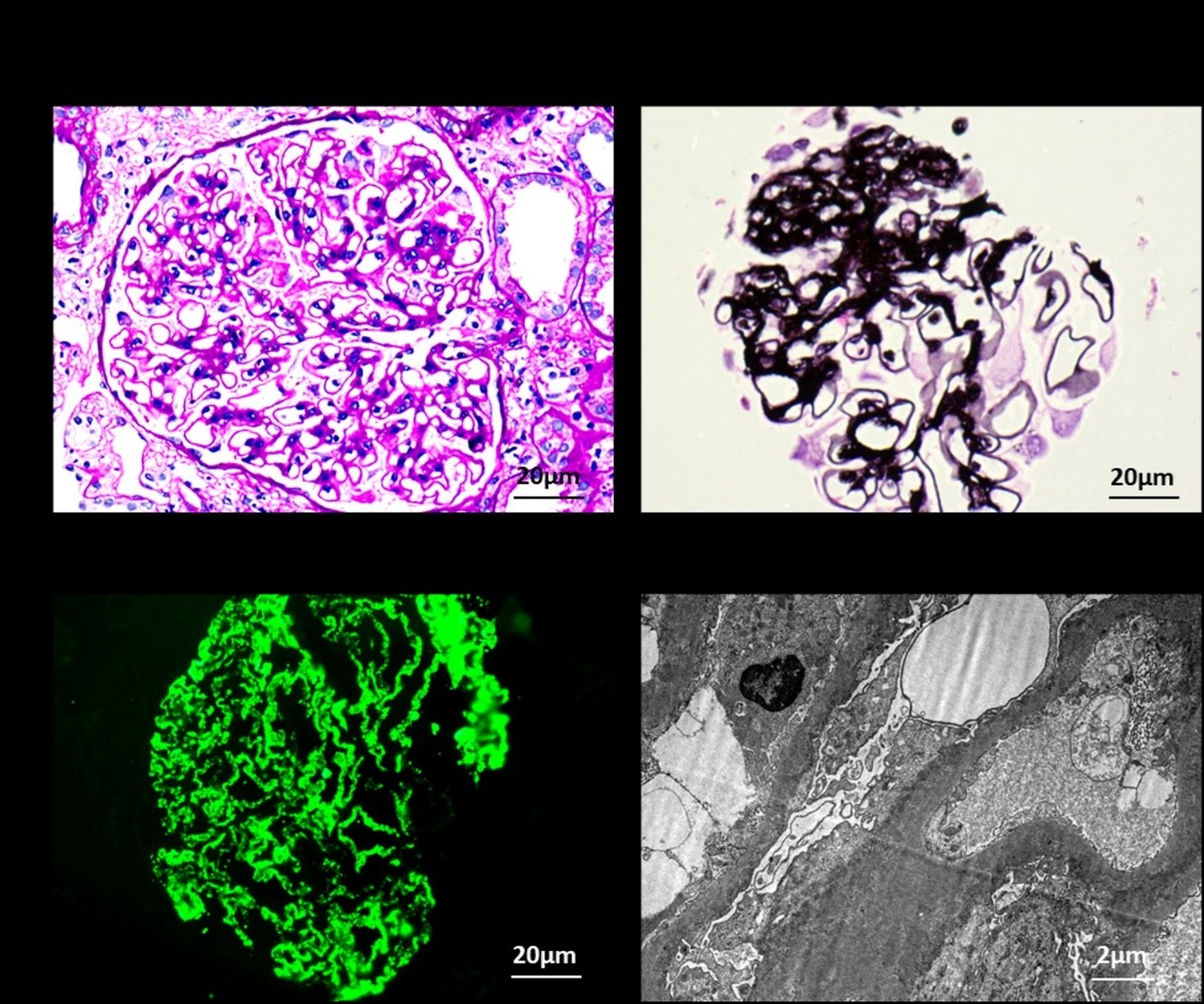
����ժҪ�������ص� 37���������ԣ���˫��֫����1�꣬����1�ܾ�� �ֲ�ʷ������������1��ǰ��ʼ��ϳ���˫��֫���ף��Խ��ײ�ˮ��Ϊ��������ĭ��ˮ�����л��⣬δ�����Ӽ���һ�����Ρ���1�ܣ����߳���˫��֫�����أ�ˮ����������ǰ�����������١�Ƥ��ؽ�ʹ���ѷ�����ʹ��Ѫ�㡢�����½����ڸɡ��۸ɵȲ��ʣ�����Ժ�������4+��Ѫ����181umol/l�������������ۺ����������ܲ�ȫ������Ժ����������������ʳ����˯�����������������С�����������������仯�� ����ʷ����С���ɥʧ��14��ʱ����˫��غ�跢��������32��ʱ����Ժȷ��Ϊ�������ۺ�������ʱ�鼡�����������ʹ�����Լ�������2�º�ͣ�á����ϸ�Ѫѹ�����IJ�������ʷ�������ҸΡ����εȴ�Ⱦ��ʷ���������ˡ�����ʷ������ҩ�ʳ�����ʷ�� ����ʷ��Ϣ��ְԱ��������������ˮ�Ӵ�ʷ���������̡�����ʷ�� ����ʷ��Ϣ��δ��δ���� ����ʷ��Ϣ����ĸ���彡����һ������ѻ���1�ӡ���ĸ���������ȷ��Ѫѹ���������ಡ�����������Լ���ʷ�� ��Ժ���壺����188cm������114.7Kg��BMI 32.18kg/m2��Ѫѹ131/83mmHg���������ף�˫��֫IIˮ�ף�˫���鷿�������Ľ���ƫ���Ρ�������ϵͳ��������ԡ���ֳ�����壺��ëϡ�裬������С��Լ5cm��˫��غ����С��ͼ1A���� ���ξ�������Ժ��������ؼ����飺�鼡��181umol/L��Ѫ���ص�8.57mmol/L��Ѫ����27.8g/L���ף�4+������������/���� 14316mg/g.Cr��24Сʱ����19g/24h�����ϸ��183/HPF�����ϸ��45/HPF���ܵ��̴�9.38mmol/l���������� 9.25mmol/l��Ѫ���桢��Ѫ�������ʡ��ո�Ѫ�ǡ��������˿����ס���������ϸ���������塢��GBM���塢�����ʪ���ӡ����塢Ѫ�̶���Ӿ�����ס����̲����ȼ�����������ؼ��ؼ�飺غͪ 0.32ng/ml������������ 0.29mIU/ml�������Խ��ͣ���Ƽ��ء���������Ƥ�ʼ��ء��������ء������ء����������صȾ������� ʮ��ͨ���ĵ�ͼ��ʾ��������ɣ������ĵ�ͼ��64���ز�CT��ʾ���������ƣ�ͼ1B�����ҷ���Ҷ�ɼ��ʵ�Խ�ڣ�������ֳ����೬��ʾ������λ�����ݸ�����λ�������Ҳ࣬�����ǻ�Ҵ�С������EF��62%�����ۣ�������λ�ģ��������Ź��ܽ��ͣ������ʳ���ʾ��֬���Σ���������δ����������������������ʾ��������13.8cm����7.8cm����̬��������Ĥ�⻬��Ƥ�ʻ������ʣ�����ϵͳδ�����롣�Ҳ�������϶ι�ǻδ�����ţ��ɼ���δ���쳣������ʾ�����׳�ӯǷ�ѣ��ɼ������ڲ�δ����ȷ��ʯ������ռλ�Ի�������ֳ������ʾ�����غ��Լ1.5��0.7cm���Ҳ�غ��Լ1.6��0.6cm��˫��غ����λ����̬��С��˫��Ա����˫�غ��̬��С������˫��Ա������Բ��CDFIʾѪ���ź�δ��ʾ�쳣������˫��غ�衢��غ����δ������ȷ���쳣ռλ�Ի�����ʾ�����ۣ�˫��غ����С������Ź�����ʾ���Դ�����̬��С�������߶�Լ0.6cm������δ����ȷ�쳣�źš��������ƫ�����ӽ�����������ѹ���������δ���쳣�����������Ը�Ҷ��̬����С���ź�δ���쳣�����ۣ��Դ����С����̬���������������ƫ��ͼ1C���� ͼ1�������ٴ����� A��������غ���С��B����λ�ģ���ɫ��ͷ����C������������ƫ����ɫ��ͷ���� ��Ժ��2�컼���ų������̾��Խ���֢����������죨ͼ2�����⾵������2��Ƥ����֯��10��������С��3������Ӳ����1���ڶ�Ӳ����������С���������ϸ����90-110��/��ϵĤϸ����ϵĤ���������������ϸ���������䣬��С���ڿɼ���ϸ������0-3��/��ëϸѪ���ȱ�����Ӳ������Ѫ���ɷֳ���������Ƿ�ѡ�3����С��Bowman��s�ұ����ֲ㣬1������ճ����PASM��MassonȾɫ����С�����Ĥ������Ƥ�¿ɼ��ȸ�����������ɼ���ͻ��δ��˫���γɡ������ʿɼ�������ĭϸ������С����Ƥϸ��ˮ�ף����������Լ����������ԣ���ǻ�ڿɼ������͡���������С��ή����������ά����������ϸ����������С���������š���������СҶ�䶯����С����δ�������쳣������ӫ��:��3����С��IgG(+++)��IgG3(+)��IgG4(++)��CLq(+-����PLA2R(++)��ëϸѪ�ܳʿ�����������Ig G1(-)��IgG2(+-)��IgA(+-)��IgM(+-)��C3(+-)��C4(-)���羵�������羵�¼�ëϸѪ����Ƥϸ�����Կ��ݱ��ԣ������ǻ�ڿɼ���ϸ���ۼ�����������Ƥϸ��������ëϸѪ���Ὺ�š���С�ұڲ����������ڲ�ϸ�����ݱ��ԣ�����������������Ĥ����������ȴ�1200nm�������Ƥϸ�����ͣ����ݱ��ԣ���ͻ�����ںϡ���Ƥ�¡�����Ĥ�ڼ�������������������ֵ�������������ڻ���Ĥ�ڣ������ձ��֡�ϵĤϸ���ͻ���������δ�������������������С����Ƥϸ�����ݱ��ԡ������������ⲡ�䡣������ϣ���ϻ��߹⾵���羵��飬�������Ϊ1.Ĥ���������� 2.����ڶ�Ӳ������С�����ף�NOS�ͣ�;������������֬øA2���壨PLA2R��������Ϊ188.63RU/ml�����ջ������Ϊԭ����Ĥ�����������������ۺ������������ۺ������������ಡ3�ڡ�����ȱ�硣 ͼ2: ����첡����� A��PASȾɫ��ʾ��С�����Ĥ����B��PASM Ⱦɫ��ʾ��С��ڶ�Ӳ����C������ӫ��Ⱦɫ��ʾ��С��ëϸѪ�����߿���״IgG������������= 20��m ��D������������ʾ����Ĥ����������ȴ�1200nm����ϸ�����͡���ͻ�������ںϡ���Ƥ�¼�����Ĥ�е����������ʳ�����������= 2��m �� ���߽�һ�����������˻����⣬�����ʾ�� ��1������ͻ������ ���� ת¼�� Ⱦɫ��λ�� �ο���� ������ ����λ�� ������仯 ͻ������ ����״̬ ������ AMER1 ENST00000330258 X:63409958 C T c.3209G>A p.Ser1070Asn ����ͻ�� ������ 100% ���Ƽ���ã� ����Ĥ�������ϲ�����������Ϊ��Σ�飬����ͨ��������ѡ����+����Ī˾���ƣ���ͬʱ���Զ�ɳ̹���ס�����Ƭ��������θ�����Ƶ��ۺ����ƣ�Ϊ��12�ܵ��ٴ������ʾ����ʵ���Ҳ����������ơ�����ˮƽ��181 ��mol/L����122 ��mol/L��24Сʱ������19�˽���3.5�ˣ���1����FK-506��Ũ��ά�������Ʒ�Χ�ڡ� ��2����Ժ����ý�� 0 �� 4 �� 8 �� 12 �� �ο���Χ ���� ��umol/l�� 181 162 136 122 57.0-97.0 ���أ�mmol/L�� 8.81 8.21 8.01 9.45 3.1-8 Ѫ����ף�g/L�� 27.8 31.6 29.6 32 40.0-55.0 �ܵ��̴���mmol/L�� 9.38 8.89 9.78 10.2 0-5.2 ����������mmol/L�� 9.25 7.3 6.5 6.44 0.0-1.7 ����Ī˾Ũ�ȣ�ng/ml�� 7.3 4.1 5.6 1.75-7.81 �� 4+ 4+ 4+ 3+ Negative ���ϸ��/ �߱��� 183 230 50 14 0-10 ���ϸ��ϸ��/ �߱��� 45 30 10 2 0-10 ��������/������mg/g.Cr�� 14316 7776 7659 5218 0.0-30.0 24Сʱ������g/24h�� 19 12 3.5 0.0-0.1 ���� �������ۺ�����KS����һ�麱��������������Ϊ�����ټ����ͷż��صIJ�����/������ȱ�ݣ����������ȱʧ��������ˡ����ۺ�������������������쳣��أ����������ѡ�������ȱ�硢�����˫����غ֢��˫�ֹ���֢�������˶�������ָ����ֺ���Σ������������쳣��[1]���ò����ٴ����Ŵ�ѧ�Ͼ��������ԣ�����40����������䷢�����ƣ�����KAL1��FGFR1��SEMA3A28���ɳʳ�Ⱦɫ�������Ŵ�����Ⱦɫ�������Ŵ���X���������Ŵ���Ŀǰ��30-40%��KS������ȷ�����ͻ�������[2]������Ϊ��ˣ�KS������������ٴ����ּ�������飬Ŀǰ��Ҫ���ٴ�������ݰ�����(1)���ٵ��£����߿ɱ���Ϊ�ڶ��������������ԣ�����ɼ���ëϡ�裬С������Сغ�裬���Ҳʳ���ʾغ�����ƫС��(2)����쳣���ɱ���Ϊ˫�ǻ��������˻�ȱʧ��(3)ʵ���Ҽ����ʾ�Լ����½��������ټ����½������������������������(4)���ֿɰ�������ϵͳ��ȱ�ݣ�������/���ѡ�������ȱ�硢�����˫����غ��˫�������˶�(�����˶�)����ָ��ֺ�����ε�����ϵͳ���쳣[3-4]�� �û�����С����˫�����ɥʧ��14��ʱ��ͬ���˷����������ڶ�����δ��������18���Ժ����������죬22��ʱֹͣ��Ŀǰ����188cm������114.7Kg��BMI 32.18kg/m2������ƫ���֣�5��ǰ����Ժ��С������Сغ�裬�ۼ��ؼ������������µͣ��ٴ����ΪKS���˴�סԺ��鸹��������ʾ�������ȱ�磬����ʾ�����λ�ģ���ֳ��������ʾ˫��غ����С�����ϻ������KS��ȷ�� Ŀǰ��֪��KS��������ı�����ҪΪ��������ȱ�磬����һЩ��������������KS���߳��ֵ����������ܵ��쳣��һƪ��������������һ������X����KS��10���ͯ�����������ۺ��������û���ͬʱ����X�������۲��ۺ���������һ������̴�������øȱ��������Ŵ���Ƥ���������̴��������Ļ��ۿ��ܸ�����С���˹�Ĥ���������ܵ��µ�������ˣ��û��ߵĵ�������������۲��ۺ����й�[5]����һ�����7�����е��������KS���ߵĻع��Է������֣���2�����߱���Ϊ�����ۺ������ҵ�����ɳ����ڸ�Ѫѹ����������֮ǰ���Ʋ���������ڵ������ർ�µľ���ڶ�Ӳ������С���������𣬵���2�����߾�δ�õ���ȷ��ϣ�Ҳ������챨��[6]�������ǵIJ����У������֤ʵ��PMN�����߱����Ϊԭ����Ĥ����������������������KS�ϲ�PMN�IJ�����ͬʱҲ��������������ڶ�Ӳ������С������NOS�ͣ���һ����Ҳ���ϼ������������Ʋ⡣ Ϊ��һ����֤����KS����ͻ�����ͼ������ۺ������Ƿ�ͬ����ͻ����أ����߽����˻����⣬�����ʾAMER1������ڴ���ͻ�䣬ͻ����Ϊ100%����1�����丸ĸ�ͽ���δ��������ͻ�䡣��ͻ�������KS��һ����ͻ�䡣��Ϊһ�������ϵ���о�̽����KS�ض��Ļ�����-���͵Ĺ�ϵ������������ȱ��������˶��������˶����ƺ���X������KS�ı��ͱ����[7]����AMER1����λ����λ��XȾɫ���ϣ���WNT/��-cateninͨ·�ĸ��������ӣ�����̥����������Ҫ���ã����ڰ�����ϵͳ������Ƥ�����ٺ�����ȶ�����֯�б��ͬKS������һ��[8-10]�������ڻ��ߵĸ�ĸ�ͽ����δ��������ͻ�䣬�������ٴ�������֧�֣�������λ�ĺ�����ȱ�磬��Щ����X������KS�ı��ͱ�������AMER1��X����ͻ�������KS�йء� ͬʱ����ȫ�������������δ�����������ۺ�����صĻ���ͻ�䣬�������ߵ�Ĥ������������һ��żȻ������KS�IJ���֢������ͬ�������˶Թ������Ŵ�������ʧ�����Ƶ��Ʋ⡣��������������ʵ���������ֻ��ⷽ�����Ч�ԣ�֧�����ڴ˱�����PMN�����߽鵼���ʡ� �������ؿ���KS�ı����ף�������������ȱ�������߽鵼��������֮������������˹ؼ����⡣��ǿ���˶�ѧ�������ں����Ŵ��ۺ����е���Ҫ�ԣ���Ϊδ���о����ڷ��������༲��֮��Ĺ�ͬ����ͨ·�ṩ�˻�������Ҫ��һ�����о�������KS��PMN֮��Ĺ�ϵ����̽��AMER1�Ȼ�������Щ�����е����á� �ο����� 1. Kumar Yadav R, Qi B, Wen J, Gang X, Banerjee S. Kallmann syndrome: Diagnostics and management. Clin Chim Acta. 2025 Jan 15;565:119994. 2. Chu G, Li P, Zhao Q, He R, Zhao Y. Mutation spectrum of Kallmann syndrome: identification of five novel mutations across ANOS1 and FGFR1. Reprod Biol Endocrinol. 2023 Mar 1;21(1):23. 3. Sonne J, Lopez-Ojeda W. Kallmann Syndrome. 2023 May 16. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2024 Jan�C. 4. Dwyer AA, Stamou MI, Anghel E, et al. Reproductive Phenotypes and Genotypes in Men With IHH. J Clin Endocrinol Metab. 2023 Mar 10;108(4):897-908. 5. Krishnamurthy S, Kapoor S, Yadav S. Nephrotic syndrome with X-linked ichthyosis, Kallmann Syndrome and unilateral renal agenesis. Indian Pediatr. 2007 Apr;44(4):301-3. 6. Duke V, Quinton R, Gordon I, Bouloux PM, Woolf AS. Proteinuria, hypertension and chronic renal failure in X-linked Kallmanns syndrome, a defined genetic cause of solitary functioning kidney. Nephrol Dial Transplant. 1998 Aug;13(8):1998-2003. 7. Stamou MI, Georgopoulos NA. Kallmann syndrome: phenotype and genotype of hypogonadotropic hypogonadism. Metabolism. 2018 Sep;86:124-34. 8. Huff V. Wilms tumours: about tumour suppressor genes, an oncogene and a chameleon gene. Nat Rev Cancer. 2011 Feb;11(2):111-21. 9. Mi J, Parthasarathy P, Halliday BJ, Morgan T, Dean J, Nowaczyk MJM, et al. Deletion of Exon 1 in AMER1 in Osteopathia Striata with Cranial Sclerosis. Genes (Basel). 2020 Nov 30;11(12). 10. Lei S, Chen C, Han F, Deng J, Huang D, Qian L, et al. AMER1 deficiency promotes the distant metastasis of colorectal cancer by inhibiting SLC7A11- and FTL-mediated ferroptosis. Cell Rep. 2023 Sep 26;42(9):113110.